
Our Research
Our researchers conduct basic research with the goal of taking their research through the translational process and putting discoveries into practice. In many cases, research is disseminated into clinical studies on diseases, disorders and conditions to improve the health and wellness of a range of patient populations.
To grow a multidisciplinary and collaborative research infrastructure that leverages intellectual capital and enhances the success of our scientists in translating discoveries to promote the advancement of human health and reducing health disparities.


Top 4%
Tier One Research - one of the nation's top 4% research institutions
$521.6M
in total sponsored program award and research activity (FY25)
72
Fulbright Scholars
15
National Academy Members
Highlights

New Center for Brain Health pairs expert care with scientific discovery
The University of Texas Health Science Center at San Antonio’s (UT Health San Antonio) new Center for Brain Health will be a transformational facility bringing clinical care, innovative research and caregiver support together under one roof to revolutionize how we understand and treat diseases of the brain. The $100 million, 103,000 square-foot center was designed to provide a comprehensive and hopeful experience for individuals facing neurological diseases, while advancing the science behind diagnosis and treatment.
Metabolic treatment: A potential breakthrough in diabetic heart disease
Heart disease is the leading cause of death for people with Type 2 diabetes and people with diabetes are more than twice as likely to experience a heart attack or stroke, according to the American Diabetes Association. While most cardiologists focus on the heart’s hemodynamic function, there is growing interest in the heart’s metabolic processes and how optimizing fuel sources may benefit individuals with heart disease.
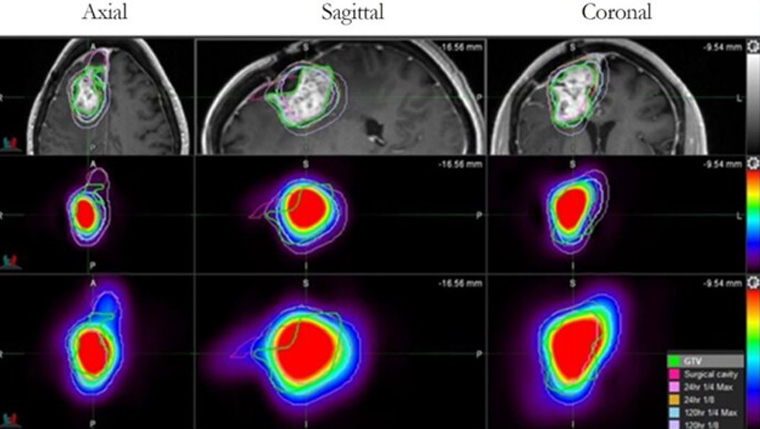
UT Health San Antonio develops drug found to more than double survival time for glioblastoma patients
A drug developed at The University of Texas Health Science Center at San Antonio (UT Health San Antonio) has been shown to extend survival for patients with glioblastoma, the most common primary brain tumor in adults.

Thirty years of excellence: San Antonio Nathan Shock Center a global leader in aging research
Over the past 30 years, scientists at The University of Texas Health Science at San Antonio (UT Health San Antonio) Nathan Shock Center of Excellence in the Basic Biology of Aging have been dedicated to the discovery and testing of novel pathways that have the potential to extend life and promote healthy aging. Recently, the center’s directors announced that they are approved for continued National Institute on Aging funding for the next five years.